This spreadsheet is a numerical simulation of absorption
spectroscopy. It computes the measured absorbance and plots the
analytical curve (absorbance vs concentration) for a simulated
absorber measured in an absorption spectrophotometer with variable
wavelength, spectral bandpass and unabsorbed stray light, given
the maximum absorptivity, path length, and half-width of the
absorber, and the slit
width and percent unabsorbed stray light of the
monochromator. The arrow buttons below each of these parameters
allow you to change the values quickly without typing. The spectra
and the analytical curve change dynamically as the variables are
changed. Any list of concentrations can be typed in for the
analytical curve. The spreadsheet fits a straight line to the
calibration curve and calculates the slope, intercept, the
correlation coefficient R2, and the
percent relative error in predicting concentrations from the
fitted line.
Alternative versions:
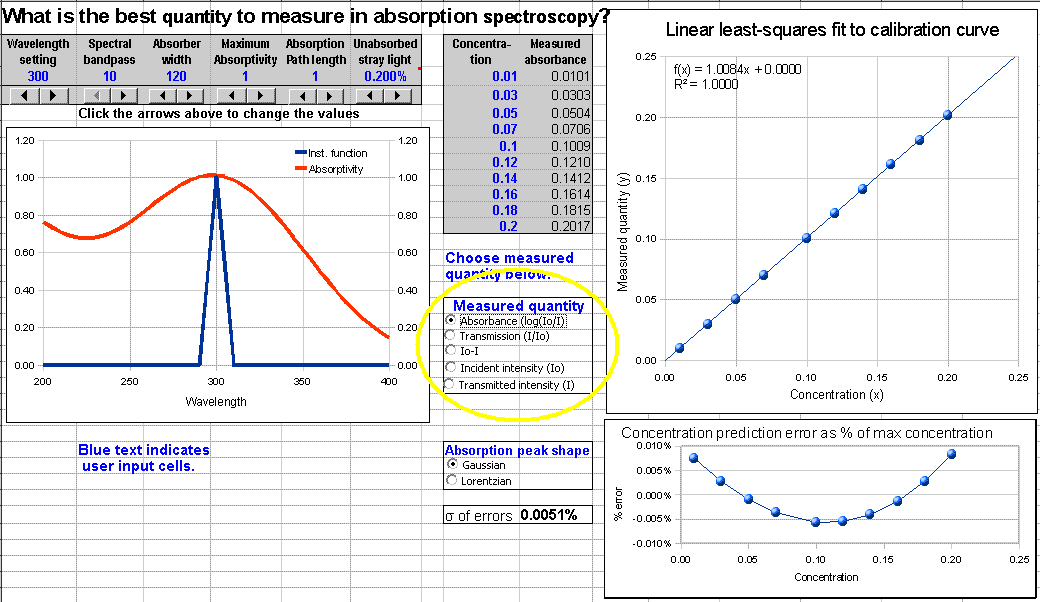
Version 1 is the basic (simplest) version. The optional Version 2 allows the user to select
which quantity to plot vs concentration: absorbance (log(Io/I)), transmission (I/Io), absorbed intensity (Io-I), or
I and Io separately. This
version can be used to demonstrate the utility of computing
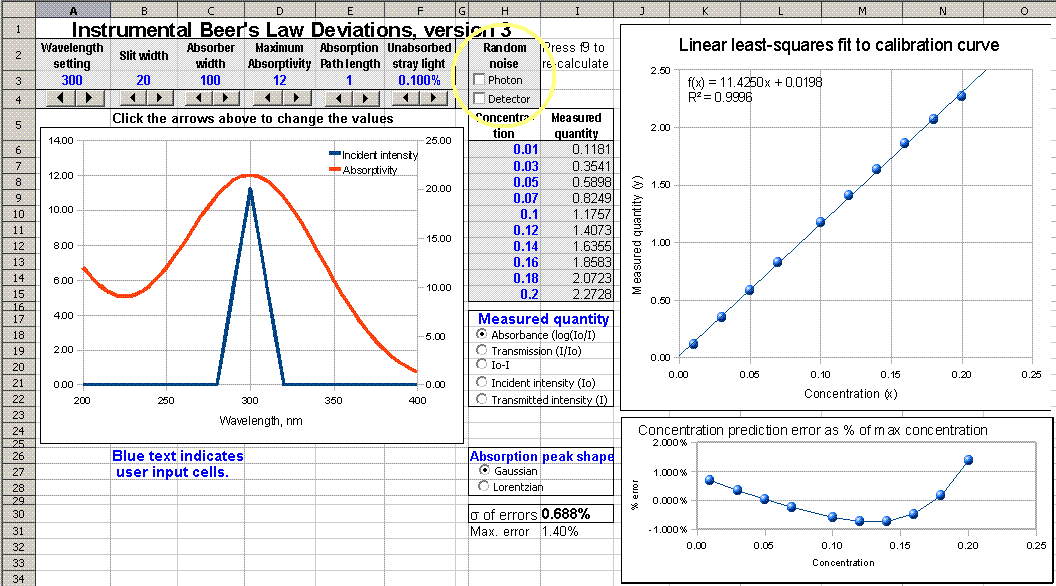
absorbance. Version 3 includes
optional random noise in the measurement of light intensity
(photon and/or detector noise), which is more realistic.
Assumptions of this simulation:
The true monochromatic absorbance is assumed to follow
the Beer-Lambert Law; the absorber spectrum consists of two peaks,
at fixed wavelengths of 150 and 300 nm, that have either Gaussian
or Lorentzian shape (selectable by user); the spectral width of
the light source is much greater than the monochromator spectral
bandpass; the monochromator has a triangular slit function (i.e.
the entrance and exit slits are equal in width and are rectangular
in shape); the absorption path length and absorber concentration
are both uniform across the light beam; the spectral response of
the detector is much wider that the spectral bandpass of the
monochromator. Only version 3 includes the effect of random noise
(see Signal-to-noise ratio of absorption
spectrophotometry,Effect of Slit Width on Signal-to-Noise
Ratio in Absorption Spectroscopy, and Comparison of Calibration
Curve Fitting Methods in Absorption Spectroscopy
for other simulations that include random noise).
Note: In the quantitative analysis of known absorbers,
these instrumental deviations from Beer's Law can be avoided
computationally by applying curve-fitting to the spectra, rather
than to the calibration curve, using the
Transmission Fitting (TFit) Method.
The Beer-Lambert Law. In
absorption
spectroscopy, the intensity I of light passing through an
absorbing sample is given by the Beer-Lambert Law:
I = Io*10-(alpha*L*c)
where “Io” is the intensity of the light incident on
the sample, “alpha” is
the absorption coefficient or the absorptivity of the absorber, “L” is the
distance that the light travels through the material (the path
length), and “c” is the concentration of absorber in the sample.
The variables I, Io, and alpha are all functions of wavelength; L and c
are scalar. In conventional applications, measured values of
I and Io are used to compute the absorbance,
defined as
A = log(Io/I)
= alpha*L*c
Absorbance defined in this way is (ideally) proportional to
concentration, which simplifies analytical calibration.
The absorption coefficient alpha
is determined experimentally. If you solve the above
equation for alpha, you
get:
alpha = A/(L*c)
So by measuring the absorbance A of a known concentration c of the absorbing compound
using an absorption path length L,
you can calculate alpha.
Because A has no units
(it's the log of a ratio of two intensities, so the intensity
units cancel out), the units of alpha are the reciprocal of the units of
L and c. For example, if the path length L is in cm and the
concentration c is in
moles/liter, alpha is in
liters/mole-cm. Even better is to prepare a series of
solutions at different concentrations and plot the measured
absorbances vs the concentrations; the resulting plot is called a
calibration curve or analytical curve. The
slope of this curve isalpha*L, so if you
measure the slope and divide by L, you havealpha.
Deviations
from the Beer-Lambert Law. It's important to understand
that the "deviations" from the Beer-Lambert
Law discussed here are not actually failures of this law but
rather apparent deviations caused by failures of the measuring
instrument to adhere to the condition under which the law is
derived. The fundamental requirement under which then
Beer-Lambert Law is derived is that every photon of light striking the detector must have an equal chance of
absorption. Thus, every photon must have the same
absorption coefficient alpha,
must pass through the same absorption path length, L, and must
experience the same absorber concentration, c. Anything that upsets
these conditions will lead to an apparent deviation from the law.
For example, any real spectrometer has a
finite spectral resolution, meaning that an intensity reading at one
wavelength setting is actually an average over a small spectral
interval called the spectral
bandpass. Specifically, what is actually measured is a convolution of
the true spectrum of the absorber and the instrument function. If
the absorption coefficient alpha
varies over that interval, then the calculated absorbance will no
longer be linearly proportional to concentration (this is called the
polychromatic
radiation effect). This effect leads to a general concave-down
curvature of the analytical curve.
Another source of instrumental non-ideality
is stray light, which is
any light striking the detector whose wavelength is outside the
spectral bandpass of the monochromator or which has not passed
through the sample. Since in most cases the wavelength setting of
the monochromator is the peak absorption wavelength of the analyte,
it follows that any light outside this range is less absorbed. The
most serious effect is caused by stray light that is not absorbed at
all by the analyte at all; this is called unabsorbed stray light. This effect also leads to
a concave-down curvature of the analytical curve, but the effect is
relatively minor at low absorbances and increases quickly at high
absorbances. Ultimately, unabsorbed stray light results in a flat
plateau in the analytical curve at an absorbance of -log(fsl), where fsl is the fractional stray
light.
There are other potential sources of deviation that are not included
in this simulation, either because they are usually not so serious
under the conditions of typical laboratory applications of
absorption spectrophotometry, or because they can be avoided by
proper experiment design. These are:
(a) unequal light path lengths
across the light beam. (In most laboratory applications, the
samples are measured in square cuvettes (sample vessels) to insure
a constant path length for all photons. When round test-tube
sample cells are used, the light beam passing through the sample
is restricted to a the central region of the sample tube in order
to minimize this effect);
(b) unequal absorber concentration across the light beam.
(Solution samples are carefully mixed before measurement to insure
homogeneity);
(c) changes in refractive index at high analyte concentration
(most analytical applications operate at lower concentrations);
(d) shifts in chemical equilibria as a function of concentration
(solutions may need to be buffered to prevent this, or the
measurement can be made at the "isosbestic
point", or a multicomponent analysis
may be performed if the spectra of all the species in equilibrium
can be determined);
(e) fluorescence of the sample, in which some of the absorbed
light is re-emitted and strikes the detector (most analytes are
not fluorescent, but if so, this error can be reduced by using a
spectrophotometer that places the sample between the light source
and the monochromator, such as a photodiode-array spectrometer);
(f) light-scattering by the sample matrix, especially in turbid
samples (this is a common source of variable background
absorption, which can be reduced by using a spectrophotometer that
places the sample cuvette right up against the face of the
detector so that it captures and detects a large fraction of the
scattered light).
(g) if the light intensity is extremely high (like a focused
laser), it's possible to observe non-linear optical effects, which
are a fundamental failure of the Beer-Lambert Law. This will
happen, for example, as the absorber approaches optical saturation (equal
populations of molecules in the ground and excited states), in
which case the sample no longer absorbs light.
The simulation here includes only the two most common
instrumental deviations from Beer's Law: polychromaticity and
unabsorbed stray light errors. The simulation operates like any
numerical integration, by slicing up the spectral range viewed by
the detector into a large number of small slices and assuming that
the Beer-Lambert Law applies over each small slice separately. The
sample absorption is represented in this simulation by a single
absorption band of either Gaussian
or Lorentzian
shape (selectable by the user) and adjustable width. The spectral
bandpass of the monochromator is represented by a triangular
function of adjustable width. Then all the separate slices are
summed up to represent the incident and transmitted light signal
measured by the detector. As it turns out, one does not need
to use very many slices to obtain a good model of the operation of a
typical absorption spectrophotometric measurement (5 nm slices are
used in this case).
The calibration curve.
In principle, it is possible to determine the concentration of
an unknown solution of by solving the above equation for
concentration:
c = A/(L*alpha)
So, you could calculate the
concentration c by
measuring the absorbance A
and dividing it by
the product of the path length L
and absorptivity alpha. That
is, if you know alpha.
Values of alpha are
tabulated for many common molecules, but the trouble is that alpha varies as a function
of wavelength, temperature, solvent, pH, and other chemical
conditions, so if the conditions of your sample don't match
those with which the alpha was measured, the calculated
concentration won't be correct. Also, the absorbances measured
on your instrument may not vary linearly with concentration, due
to the deviations discussed above, in which case no single value
of alpha would give
accurate results. As a result, it is much more common in
practice to prepare a series of standard solutions of known
concentration, whose chemical conditions as close as possible to
those of the sample, measure their absorbances on your
instrument, and plot a calibration
curve with concentration of the standards on the x-axis
vs measured absorbance on the y-axis. (If Beer's Law is
observed, the slope of this curve isalpha*L). Once the
calibration curve is established, unknown solutions can be
measured and their absorbances converted into concentration
using the calibration curve. Here is a
graphic animation of this calibration process. This can
be done either graphically (by drawing a line from the
absorbance of each unknown across to the calibration curve and
then down to the concentration axis) or it can be done
mathematically (by fitting a line or curve to the calibration
data, solving the equation of that line for concentration, then
using that equation to convert measured absorbances to
concentration). With computers, it's usually easier to do the
latter. (See "Comparison
of Calibration Curve Fitting Methods in Absorption
Spectroscopy" to see how to fit non-linear calibration
curves). The important point is that even if Beer's Law is not obeyed, you can get
accurate resulting using a calibration curve.
Student handout for OpenOffice version.
Instrumental Deviation from Beer's Law
1. Open http://terpconnect.umd.edu/~toh/SimpleModels/BeersLaw.ods in OpenOffice Calc (August
6, 2008 version or later). This spreadsheet simulates an
optical absorption spectroscopy measurement and demonstrates how
the instrument's measurements of absorbance can deviate from the
ideal predicted by the Beer-Lambert Law (a.k.a. Beer's Law).
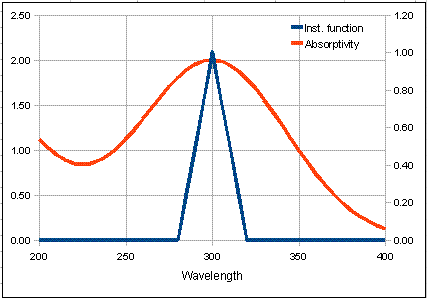
The graph on the left
of the window shows the absorption spectrum of the analyte in red over a wavelength range from
200 - 400 nm. The blue
line is the "Transmitted intensity" ; it shows the
spectrum of light emerging from the exit slit of the
monochromator and passing through the absorbing sample. Despite
its name, a monochromator never really passes a single color or wavelength
of light; it actually passes a small range of wavelengths. This range of
wavelengths is called the "spectral bandpass". The smaller
the slit width, the smaller the spectral bandpass, and the more
nearly monochromatic is the light emerging from the exit slit.
In normal laboratory instruments, the spectral bandpass is
controlled by the slit
width, which is adjustable by the experimenter on
many instruments (but not on the Spectronic
20, which has a fixed 20 nm spectral bandpass). In this
simulation you can vary the slit width of the simulated
instrument from 10 nm to 100 nm by using the Slit width control above the
graph, but it can not be set below 10 nm (every instrument has a
minimum slit width, and therefore a minimum spectral bandpass
setting; you can not set the slit width to zero because then no light would get in and
the instrument would not work at all! Note that the transmitted
intensity has a triangular spectral distribution (because the
entrance and exit slit widths are always equal in a normal
monochromator.
The peak of the slit
function falls at the wavelength setting of the monochromator.
You can control the wavelength setting by using the Wavelength setting control
above the graph; this is equivalent to turning the wavelength
knob on the spectrometer.
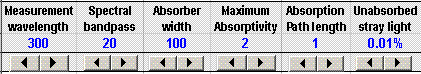
The other controls above the
graph are for the other variables in this simulation, such as the
path length of the absorption cell (1-10 cm). So that you
can see how different types of absorbing species would behave, the
simulation allows you to vary the maximum absorptivity of the
analyte and the spectral width of the absorber (that is, the width
of the absorption bands that constitute the absorber's spectrum).
The last control is for the stray light. Every real
monochromator passes a small amount of white light as a result of
scattering off optical surfaces within the monochromator (mirrors,
lenses, windows, and the diffraction grating). Usually this
so-called "stray light" is a very small fraction of the light
intensity within the spectral bandpass, but it's important because
it can lead to a significant source of deviation from Beer's Law.
In most cases the monochromator is tuned to the wavelength
of maximum absorption of the analyte, in order to achieve the
greatest sensitivity of analysis. But that means that stray
light is less absorbed than the light within the spectral
bandpass. The worst offender is stray light that is not at
all absorbed by the analyte - "unabsorbed stray light", usually
expressed as a percentage of the light intensity within the
spectral bandpass. In the simulation, this is set by
the "Unabsorbed stray light" control. Typical
monochromators have stray light rating in the 0.01 - 1% range,
depending on the wavelength setting and the type of light source
used. The stray light is always worse at wavelengths where
the light source is least intense and where the detector is least
sensitive. (However, in this simulation, the stray light does not
automatically change with wavelength). Note: when adjusting
the stray light, use the number spinner (small arrows below the
number) rather than typing directly into cell F3. The other
variables you can change either by typing or by using the number
spinners.
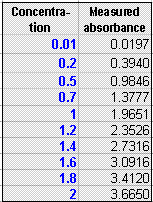
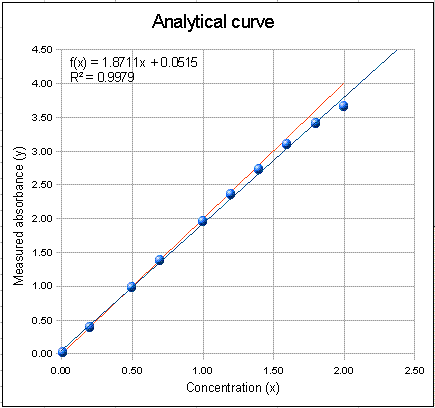
The graph on the right of the
window is the analytical curve (calibration curve), showing the
absorbances measured for each of the standard solutions listed in
the table in the top middle of the window. You can type any
set of concentrations in the concentration column of this table,
up to a maximum of 10 standards. The red line in the plot
(sometimes obscured by the other lines) represents the ideal
Beer's Law absorbances, the blue dots represent the measured
absorbances for each standard solution, and the blue line is the least-squares straight-line
fit to the concentration-absorbance data. Ideally, the
fitted straight line (blue line) should go right through the
middle of the blue dots. Also on the plot is the equation of
the fitted line (x = concentration and f(x) = absorbance) and the
R2 value, which is a measure of the
degree of correlation between absorbance and concentration (1.0000
means perfect correlation; anything less than 1.0000 is not
perfect).
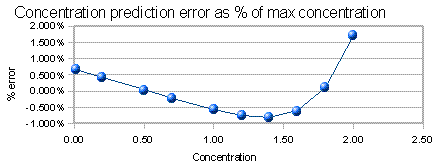
The graph below the
calibration curve is the concentration prediction error. If you
were to run the standards as unknowns and predict their
concentrations from the straight-line fit to the calibration
curve, this would be the error in prediction, expressed as a
percentage of the highest concentration. (The standard deviation
of those errors is a good single-number summary of those errors;
it is displayed to the left). This is a more sensitive indicator
of non-linearity than the R2 value.
2. Start the experiment with
a nearly ideal case (with
the spectral bandpass much less than the absorption width and no
stray light). Set wavelength = 300 nm, slit width = 10 nm,
absorber width = 200 nm, maximum absorptivity = 1, path
length = 1 cm, and unabsorbed stray light = 0. Note that the
ideal absorbances (red line), the measured absorbances (blue
dots), and the least-squares fit (blue line) are essentially
identical, even at the highest concentrations, and the R2
is exactly 1.0000, showing that the instrument readings
follow Beer's law in this nearly ideal case. You can see
that in this case the absorption spectrum is almost flat over the
spectral bandpass. This means that all the photons have
essentially the same absorption coefficient, a fundamental
requirement of Beer's Law. The concentration prediction error (the
graph below the calibration curve) is so small it is negligible
compared to other errors that are likely to be greater anyway,
such as volumetric calibration accuracy and precision. But
real-world absorption measurements are never so perfect.
3. Unabsorbed stray light limit only. Leave the
absorber width = 100 nm, slit width = 10 nm, maximum absorptivity
= 1, path length = 1 cm, and set the unabsorbed stray light =
0.1%, using the number spinner - small arrows below the number -
rather than typing directly into cell F3. For comparison, try a
stray light of 1% and 0.01% and observe the calibration curve
shape. Notice that the measured absorbance bends off from a
straight line at the highest concentrations, but still very linear
at lower concentrations. Why does the calibration curve
flatten out at high concentrations? Simple! As the
concentrations increases, the intensity of the transmitted light
from the spectral bandpass decreases towards zero, but the
unabsorbed stray light remains at the same intensity because it is
unabsorbed. So eventually at very high concentrations, all that
remains in the transmitted light is stray light, which results in
an transmittance reading of T = (I+straylight)/(Izero +
straylight), which approaches (straylight)/(Izero + straylight) as
I approaches zero. See if you can devise a rule that will
predict the plateau absorbance for a given stray light percent.
4. Typical situation in solution spectrophotometry. Set
wavelength
= 300 nm, slit width = 20, absorber width = 100, and leave
maximum absorptivity = 1, path length = 1 cm, % stray light =
0.01% Note that analytical curve plot is almost perfectly linear
(correlation coefficient is 1.0000) up to a measured absorbance of
2, yet the slope is 2% less than the ideal line (in red). In other
words, just because the the analytical curve seems to be linear
does not mean that the measured absorbance equals the ideal peak
absorbance. (Of course, in most cases you don't really need to
know the true peak absorbance, because almost all practical
applications of absorption spectroscopy in chemical analysis are
calibrated by using standard solutions). The concentration
prediction error, based on a linear fit, is less than 0.05%.
This gives an idea of the error that is caused by the slight
residual non-linearly of the calibration curve.
5. Effect of changing the wavelength. Leave
everything as it was, except return the the maximum
absorptivity to 1.0 and the stray light to 0.01%. Increase
the wavelength setting to 350 nm and see what happens: the
calibration curve plot has a lower slope, of course, because the
absorptivity is less at 360 nm that at 300 nm. But that's
not all. The curve is also substantially less linear: the R2
drops to 0.9998 and the concentration prediction error
goes up about 10-fold to
0.5%. Why should the calibration curve be less linear? Think about
the total change in the absorptivity of the analyte over the
spectral bandpass (look at how much the red line changes under the
blue triangle). When the wavelength is set at a maximum (or a
minimum), the total change in absorptivity over the spectral
bandpass is less than when the wavelength is set to the side of a
band, where the rate of change of absorptivity with wavelength is
greatest. Then think about the requirement that all the photons
have essentially the same absorption coefficient. This
effect is called the "polychromatic light" effect. You can
decrease the polychromatic light by decreasing the spectral
bandpass (using a smaller slit width).
Note that the R2 is
not a very sensitive indicator
of non-linearity: even when it is just slightly less than 1.000,
significant non-linearity may be present. Looking at the
concentration prediction error plot (also called the "residual"
plot) is more informative that just looking at the R2 value.
You might ask why some
spectrometers even have adjustable slit widths, when the best
linearity and adherence to Beer's Law is observed at the smallest
spectral bandpass. Why not just use the smallest slit width
setting all the time? The answer is that wider slits let in more
light, which improves the precision of light intensity
measurement. See the simulation "Effect of Slit Width on
Signal-to-Noise Ratio in Absorption Spectroscopy" at http://terpconnect.umd.edu/~toh/models/AbsSlitWidth.html
for a simulation of this aspect.
6.
Measuring higher concentrations
at alternative wavelengths. Suppose we wanted to measure
some high concentrations, above the usual linear range of the
calibration curve, without diluting the samples (which would be
time-consuming and possibly expensive and error prone) and without
using shorter path length cells (which also involves extra cost).
To illustrate this problem,
you can simply increase the maximum absorptivity from 1.0 to 2.0,
which will instantly double all the absorbances. Leave
absorber width = 100, source width = 20, % stray light = 0.01 and
increase the maximum absorptivity to 2.0. With the
wavelength set to the maximum at 300 nm, the linearity is not so
great (R2 = 0.998; concentration
prediction error = 0.78%). This is mainly because of stray light,
which effects the absorbance above 3.0.
What about changing the
wavelength of measurement to a less sensitive wavelength. Changing
the wavelength is quick and doesn't cost anything. But we
found in #5 that measuring on the side of a band leads to a great
increase in non-linearity. Set the measurement wavelength to 350
nm. This reduces the absorptivity (sensitivity) by about half. The
linearity in this case is actually improved (R2 =
0.9992; concentration prediction error = 0.48%) despite the fact
that the polychromatic light effect is worse at this wavelength,
as you observed in #5. That's because the stray light effect is
lessened by the reduced absorbance at the higher wavelength. In
this case the stray light effect is greater than
the polychromatic light effect.
But you can do even better
than this. In this particular simulation, the absorber has a minimum in its absorption
spectrum at about 225 nm. At that wavelength we have a similar
sensitivity reduction, which reduces the stray light effect, but
the polychromatic light effect is much less on the minimum
than on the side of the sloping slide of the spectrum. Set the
measurement wavelength to 225 nm. You can see that the linearity
is greatly improved (R2 = 1.000;
concentration prediction error = 0.023%). So the best approach is
to use the peak wavelength for lower concentrations and the
minimum as the alternative wavelength for higher concentrations.
7. Atomic absorption. Sometimes it is not possible
or practical to have the ideal situation where the spectral
bandpass is much narrower than the spectral width of the
absorption. For example, in line source atomic absorption
spectroscopy, the effective spectral width of the light source is
set by the line width of the hollow cathode lamp (not by the monochromator's
spectral bandpass), and the absorber width is determined mainly by
the temperature and pressure in the atomizer. As a result, the
absorber width is only about 3 times larger than the spectral
width of the light source. For example, the line width of the
hollow cathode lamp might be 0.001 nm and the absorber width
might be 0.003 nm. To simulate this situation, we'll let 10 units
represent 0.001 nm and set the slit width = 10, absorber
width = 30 (because it's really only the ratio of the widths that is important), stray
light = 0.1%, and change the absorption peak shape to Lorentzian
(a better match to the shape of an atomic absorption line in an
atomic absorption atomizer).
With these settings, the
measured absorbance is 6% less than the true value, but the
linearity is fairly good (R2 =
0.9999) up to an absorbance of about 2 and the concentration
prediction error is only about 0.3%.
But the situation is
substantially worse if one attempts to do continuum-source atomic
absorption with a
medium-resolution spectrometer. In that case the spectral bandpass might be 10 or
more times larger than
the absorption width. Set the absorber width = 10, slit width
= 100, and leave the peak shape set to Lorentzian. Note the
linearity is substantially worse: (R2 =
0.97; concentration prediction error = 5.6%). This is one reason
why continuum-source atomic absorption utilizes high-dispersion
"echelle" spectrometers that can achieve a spectral bandpass about
10 to 100 times smaller than conventional monochromators at the
same slit width. Note:
there are other spectroscopic complexities with line-source atomic
absorption: see Spectroscopic
Simulation of Atomic Absorption for a more specific
simulation of atomic absorption.
8. Extensions and next steps. Several extensions of
this line of investigation might be taken:
a. The
simulation
"Effect of Slit Width on
Signal-to-Noise Ratio in Absorption Spectroscopy" considers
how the slit width and the dispersion on the monochromator effects
the precision and signal-to-noise ratio of intensity and
absorbance measurement. b. "Signal-to-noise ratio of absorption
spectrophotometry" provides a more detailed model of a
UV-visible absorption spectrophotometer with a continuum source
(e.g. tungsten incandescent lamp, modeled as a blackbody),
dispersive monochromator, and a photomultiplier detector.
d. Two simulations
consider the extension to multi-wavelength data such as
would be acquired on diode-array, Fourier transform, or
computer-automated scanning spectrometers:
"The TFit Method
for quantitative absorption spectroscopy", located athttp://terpconnect.umd.edu/~toh/spectrum/TFit.html, describes a
computational approach that eliminates the calibration curve
non-linearity by basing the measurement of absorbance on a
model of the actual spectroscopy of the measurement, rather
than assuming that the instrument adheres to the limiting
case of zero spectral bandpass and zero stray light.
Frequently
Asked Questions (taken from
actual search engine queries)
1. Exactly
what does it mean to 'follow Beer's Law'?
Basically it means that the measured absorbance
is proportional to the concentration of the
absorber, that is, a plot of absorbance vs absorber
concentration is a straight line. Absorbance is defined
as log(Io / I), where
"Io" is the intensity of the light
incident on the sample and "I" is the intensity of
the light transmitted through the sample,
2.What is the equation for absorbance vs
concentration?
A
= alpha*L*c
where alpha is the absorption
coefficient (or absorptivity), L is the path length of the light
through the absorber, and c is the concentration of the
absorber. The absorbance A is defined as log(Io
/ I), where Io is the
intensity of light beam that strikes the absorber
and I is the intensity of light beam after it
passes through the absorber. This is called the Beer-Lambert Law
or Lambert-Beer Law or Beer-Lambert-Bouguer
Law. (Strictly speaking, Beer's Law refers to the relationship
of absorbance and concentration and Lambert's Law
refers to the relationship of absorbance and path length, but
the two are usually combined into one).
3. Why does
absorbance increase with concentration?
Because the higher the concentration, the more absorbing
molecules are in the light path to absorb the light. It's like
brewing tea: weak tea has a low concentration of tea dissolved
in the hot water and a light color (does not absorb much
light). Strong tea has a high concentration of
tea and a darker color (absorbs lots of light).
4. What is a Beer's Law calibration curve? How do you make
and interpret a Beer's Law plot? Why are most calibration curves of Beer's Law
rather than Lambert's Law?
The usual Beer's Law plot is a plot of concentration of
absorber on the x (horizontal)
axis, vs measured absorbance on the y (vertical) axis.
This is useful when you want to determine the concentration of
solutions by measuring their absorbance. The slope of this plot
is the product of the path length L times the absorption
coefficient alpha
where the slope
is defined as the ratio of the y-axis difference
to the x-axis difference between any two points on the line. (This
is in contrast to a Lambert's
Law plot of path length on the x axis vs measured
absorbance on the y
axis. This might be useful if you want to
determine the path length of an absorber by measuring its
absorbance. The slope of that plot would be the
product of the absorptivity alpha
times the absorber concentration c).
5. What are the
units of the absorption coefficient, alpha?
It depends on the units of concentration and path length. If
concentration is measured in moles/liter (molarity) and path
length in cm, then the units of the absorption coefficient (also
called the molar absorptivity)
are liters/mole-cm. If
concentration is measured in grams per liter and path length in
cm, then the units of the absorption coefficient
are liters/gram-cm. If
concentration is measured in grams per mL (cubic centimeters)
and path length in cm, then the units of the
absorption coefficient are mL/gram-cm.
6. How do you know the
value of the absorption coefficient, alpha?
Absorption coefficients are determined experimentally and are
tabulated for a large number of compounds in chemistry reference
works. If you solve the above equation for alpha, you get:
alpha
= A / (L*c)
So by measuring the absorbance A
of a known concentration c
of the absorbing compound using an absorption path length L, you can calculate alpha. Because A has no units (it's the log
of a ratio of two intensities, so the intensity units cancel
out), and because L and c are in the denominator, the units of alpha are the reciprocal
of the units of L and c.
Absorption coefficients vary widely from substance to
substance and also vary with wavelength. Values
of alpha are tabulated
in the literature and in reference books for many common
molecules.
7. What's the
difference between 'absorption' and 'transmission'?
Absorption refers to how much light is lost when passing through
an absorber. Transmission refers to how much light remains after
it passes through. Absorption is expressed as the absorbance, log(Io
/ I) or as the absorption, (Io-I)
/ Io, or the percent absorption, 100(Io-I)
/ Io. Transmission is
expressed as the transmittance, I
/ Io, or as the percent transmission, 100(I
/ Io). As the
absorber concentration goes up, the absorbance and
the absorption both go up, but the transmission goes
down. Of these, absorbance is the most widely used
because it is directly proportional to concentration, according
to Beer's Law. Note that all of these measures are based on the
RATIO of the two intensities Io and I.
This has the huge advantage of making these measures independent of the overall
intensity of the light source and of the sensitivity of the
detector used to measure the intensity. This in turn helps to
make these quantities independent of the instrument used to
measure it.
8. Why does the Beer-Lambert Law lead to absorbances
above 1?
An absorbance of 1 simply means that the transmitted intensity,
I, is one-tenth of the incident intensity, Io.
If the transmitted intensity is lower than that, the absorbance
is higher than 1. You might be confusing absorbance with absorption. As the absorber
concentration goes up, the absorbance, log(Io
/ I), and the absorption, (Io-I)
/ Io, both go up
(and the transmission goes down), but the absorption can't get
any higher than 1, whereas the absorbance keeps going up
proportional to concentration. You can easily get absorbances
above 1, even up to 3 or 4 under ideal conditions.
9. How do you
measure unknown concentrations with absorption
spectrophotometry? Is it better to use a standard curve or the
equation for Beer's Law?
If you solve Beer's Law for concentration, you get:
c = A / (L*alpha)
So, you could determine the concentration c simply by measuring the
absorbance A and
dividing it by
the product of the path length L and absorptivity alpha. That is, if you know alpha. Values of alpha are tabulated for
many common molecules, but the trouble is that alpha varies as a
function of wavelength, temperature, solvent, pH, and other
chemical conditions, so if the conditions of your sample don't
match those with which the alpha was measured, the calculated
concentration won't be accurate. Also, some spectrophotometers
do not follow Beer's Law exactly; it's not uncommon for some
instruments to give absorbance readings that are a little too
low and slightly non-linear with respect to concentration.
Because of this, it's better to prepare a series of standard solutions of
known concentration, made up so that the chemical conditions
are as close as possible to those of the sample, measure their
absorbances on your instrument, and plot a calibration curve with
concentration of the standards on the x-axis vs measured
absorbance on the y-axis. Once the
calibration curve is established, unknown solutions can be
measured and their absorbances converted into concentration
using the calibration curve. Here is a graphic
animation of this calibration process applied to a
specific assay. This can be done either graphically (by
drawing a line from the absorbance of each unknown across to
the calibration curve and then down to the concentration axis)
or it can be done mathematically (by fitting a line or curve
to the calibration data, solving the equation of that line for
concentration, then using that equation to convert measured
absorbances to concentration). With computers, it's usually
easier to do the latter. The important point is that even if Beer's Law is not obeyed
perfectly, you can still get accurate resulting using a
calibration curve.
10. How do you solve Beer's Law for transmittance?Does
graphing transmission or absorbance result in a more
accurate standard curve? Why is a straight line calibration
better?
If you state Beer's Law as I = Io*10-(alpha*L*c),
then just divide both sides of the equation by Io, the
result is I / Io
= 10-(alpha*L*c)
= 10-A, where A is absorbance. The quantity I /
Io is defined as transmittance. Absorbance
A is defined as log(I / Io).
In principle, either transmittance
or absorbance would work equally well for
quantitative analysis, because there is exactly the same
amount of information in an transmission reading as in an
absorbance reading; one can be converted to the other without
loss. However, a calibration curve plotted in absorbance is
linear, according to Beer's Law, whereas a calibration
curve plotted in transmission would be highly
non-linear (exponential, in fact). It's just easier to fit a
line to a set of straight-line data, and to see when the data
are deviating from that straight line, that to a fit a curved
line to non-linear data, whether it is done by hand or with a
calculator or computer.
11. What are the limitations of beer's law?
What limits the linearity of Beer's Law plot?
Deviations from Beer's Law can be caused by:
(a) Stray light, which is any light
striking the detector whose wavelength is outside the
spectral bandpass of the monochromator or which has not
passed through the sample;
(b) Polychromatic light effect,
which occurs if the absorber's absorption coefficient alpha varies over the
wavelength interval of light passing through the sample;
(c)
unequal light path lengths across the light beam;
(d)
unequal absorber concentration across the light beam;
(e)
changes in refractive index of the solution at high analyte
concentration;
(f) light-scattering by the sample matrix, especially in
turbid samples, resulting in a significant absorption
signal even when the absorber's concentration is zero;
(g)
shifts in chemical equilibrium involving the absorber as a
function of concentration;
(h) changes in pH as a function of concentration.
(i)
fluorescence of the absorber, in which some of the absorbed
light is re-emitted and strikes the detector;
(j)
chemical reactions caused by the absorption of light,
including photolysis, dimerization,
polymerization, and molecular phototropism (change in
molecular shape when the molecule absorbs light).
(k) if the light intensity is extremely high (like a focused
laser), it's possible to observe non-linear optical effects,
which are a fundamental failure of the Beer-Lambert Law.
The most common of these are (a) and (b), which both result in
a concave-down curvature of the Beer's Law plot; (c) and (d)
are easily avoided by proper experiment and instrument design
(square cuvettes, well-mixed solutions); (e) is only a problem
at very high concentrations; (f) is pretty common in
real-world applications to complicated samples, but can be
minimized by special measurement techniques and instrument
designs; (g) and (h) can be avoided by buffering
the solutions to constant pH and adjusting the concentration
of reagents; (i) and (j) occur rarely with some particular
absorber molecules and must be treated on a case-by-case
basis; (k) never occurs in standard laboratory instruments
with conventional light sources..
12. Under what
conditions is the Beer-Lambert law not obeyed?
The Beer-Lambert Law will not be obeyed if the photons of
light striking the detector do not all have an equal
chance of absorption by the sample. This can happen if
they have different absorption coefficients, different path
lengths through the sample, or if they encounter different
concentrations of sample molecules. Also if anything else is
present in the sample that absorbs light or causes light
scattering, the measured absorbance will not be zero when the
analyte's concentration is zero, contrary to Beer's Law. If
the absorber undergoes any type of chemical reaction or
equilibrium that varies as a function of concentration, Beer's
Law will not be obeyed with respect to the overall or total
concentration, because the concentration of the
actual absorbing molecule is not proportional to the overall
concentration of the solution. The "c" in Beer's Law refers to
the concentration of just the absorber, not to the total
concentration of all the compounds reacting with or in
equilibrium with the absorber. Even if Beer's Law holds
exactly for each individual compound, the total absorbance of
the mixture will not follow Beer's Law with respect to the
total concentration if the proportion of each compound changes
with concentration (unless by chance the absorptivity of all
those compounds happens to be exactly the same).
13. Why are
measurements taken in increasing order of concentration when
using spectrophotometry?
Mostly it's just a convention. Actually, the measurements can
be taken in any order; if the instrument and the samples and
standards are stable with time, the result will be essentially
the same. The one situation where the order of
measurement is done from lowest to highest concentration is if
the sample cuvette is difficult to clean thoroughly once it is
exposed to high concentrations.
14. What happens
to the energy of the absorbed light be measured? What
about the "Law of Conservation of Energy"?
Conservation of energy still works. The energy of the absorbed
light is converted into heat,
which increases the temperature of the measured samples
slightly. But in an ordinary instrument the temperature
increase is very small and not even easily measurable.
15. How can the
intensity of absorbed light be measured?
In absorption spectrophotometry the absorbed light intensity
is not measured directly, rather it's measured indirectly by
measuring the difference between the incident and transmitted
intensity.
16. I see how the detector measures the transmitted
intensity, I. How does it measure the incident intensity,
Io?
If you remove the sample from the light beam, the detector
then measures the incident intensity, Io, because
there is noting in the beam to absorb light (except air). But
for the measurement of solution samples contained in sample
cells (cuvettes),
there is an additional complication: the cuvette itself
reduces the light intensity, by light reflection from the
surfaces of the glass, with the result that an empty cuvette
would give a significant absorption signal. Also, sometimes
the solvent absorbs some of the light. To compensate for both
of these effects, you need to measure a cuvette
filled with solvent (which is called the "blank"),
and subtract the absorbance of the blank from all the
standards and samples. This effectively subtracts out the
absorption of the cuvette and solvent, and the resulting
difference is the absorbance of the analyte alone.
17. How do you use spectrophotometry to measure
things that are colorless? It
is very common in analytical spectrophotometry to use a "color
reagent" that will react with a colorless analyte under
appropriate conditions to produce a stable colored product
that absorbs in the visible, preferably at a wavelength where
other components in the sample do not absorb. There are a
large number of such reagents commercially available to meet
many analytical requirements. Another possibility
is that many colorless compounds absorb in the ultra-violet
(uv) region from 200 nm - 400 nm. If the other components in
the samples do not absorb significantly in the uv, a
spectrophotometric analysis in the uv region possible. But to
do this you must use a uv-visible spectrometer, and cuvettes
(usually quartz or fused silica) which are transparent in the
uv. As a solvent, water is quite transparent in the uv,
but if you must use another solvent, make sure its uv
absorption at the analytical wavelength is not too much.
18. Why does the Beer-Lambert Law require
monochromatic light? Actually
the Beer-Lambert Law requires that all the photons
of light striking the detector have an equal chance of absorption by
the analyte. This requires that all the photons have
the same absorption coefficient, which will be the case either if they all have
the same wavelength (i.e. monochromatic light) or if the sample
absorption is constant over the wavelength range of the light
beam (e.g. at the maximum or minimum of a broad absorption
peak).
19. What is the
low concentration limit of Beer's Law?
There is no fundamental low concentration limit, but at very
low concentrations, the readings of absorbance can be in error
due to the limited resolution of the readout display or
because of the signal-to-noise ratio of the light intensity
measurement (due to detector nose, photon noise, or light
source fluctuation).
20. What is the
high concentration limit of Beer's Law? What is the
approximate concentration above which deviation from Beer's
Law first become apparent?
Normally, above whatever concentration produces an absorbance
of about 2, deviations start to become apparent. Stray light
especially becomes more important at high
absorbances. Low-quality instruments, especially
when operated near their wavelength limits, exacerbate the
non-linearity at high absorbances.
21. What is the instrument measurement range of
transmission in absorption spectrophotometry? What
is the optimum transmittance range for optimum precision?
The measurement range of good-quality
instruments is typically from an absorbance of about
0.0001 (transmittance = 0.9998 or 99.98 %T) to an absorbance
of about 4 (transmittance =
0.0001 or 0.01%). The best precision of concentration
measurement occurs at about an absorbance of 0.5 to 1.0 (10 to
30%T), depending on what exactly is the dominant source of
random noise. This is demonstrated in the simulation Effect of Slit Width
on Signal-to-Noise Ratio in Absorption Spectroscopy.
22. Why are absorbance readings taken at the peak
wavelength of maximum absorbance? Must you always
use the maximum? What is the wavelength of least error?
It really depends on what is the largest source of error.
Taking the readings at the peak maximum is best at low
absorbances because it gives the best signal-to-noise ratio,
which improves the precision of measurement. If
the dominant source of noise is photon noise, the precision of
absorbance measurement is theoretically best when the
absorbance is near 1.0. So if the peak absorbance is below
1.0, then using the peak wavelength is best, but if
the peak absorbance is well above 1.0, you might
be better off using another wavelength where the absorbance is
closer to 1. Another issue is calibration curve
non-linearity, which can result in curve-fitting
errors. The non-linearity caused by polychromatic
light is minimized if you take readings at either a peak
maximum or a minimum, because the absorbance change with
wavelength is the smallest at those wavelengths. On the other
hand, using the maximum increases the calibration
curve non-linearity caused by stray light. Very
high absorbances cause two problems: the precision of
measurement is poor because the transmitted
intensity is so low, and the calibration curve linearity is
poor due to stray light. The effect of
stray light can be reduced by taking the readings at a wavelength where the absorbance is lower
or by using a non-linear
calibration curve fitting technique. Finally, if
spectral interferences are a problem, the best measurement
wavelength may be the one that minimizes the relative
contribution of spectral interferences (which may or may not
be the peak maximum). In any case, don't forget: whatever
wavelength you use, you have to use the exact same wavelength for all
the standards and samples.
23. How can you identify the cause of the deviation
from Beer's Law?
There are two experiments you can perform that will throw at
least some light on this question (pardon the
pun). First, measure the absorbance of a single
concentration at different path lengths (by using different
sample cuvettes) and plot the measured absorbance vs path
length (this is a Lambert's Law plot). Second, prepare a
series of standard solutions of different concentrations,
measure then in a fixed path length, and plot concentration vs
absorbance (this is a normal Beer's Law plot).
Fit a straight line to both of these sets of data.
If the Lambert's Law plot is
non-linear (concave down),
then the problem is optical rather than chemical, most likely
polychromatic light or stray light. If
the plot is linear at low concentrations but non-linear at
high concentrations, it's probably stray light. If the
Lambert's Law plot is linear, but the Beer's
Law plot is non-linear, it suggests that the
non-linearity is chemical in nature, perhaps an equilibrium
shift that depends on the concentration of the solution.
24. How
can you distinguish between random and systematic
deviation from Beer's Law? How can I locate the
non-linear region?
Fit a straight line to the calibration data and look at a plot
of the "residuals", the differences between the y values in
the original data and the y values computed by the fit
equation. Deviations from linearity will be much
more evident in the residuals plot than in the calibration
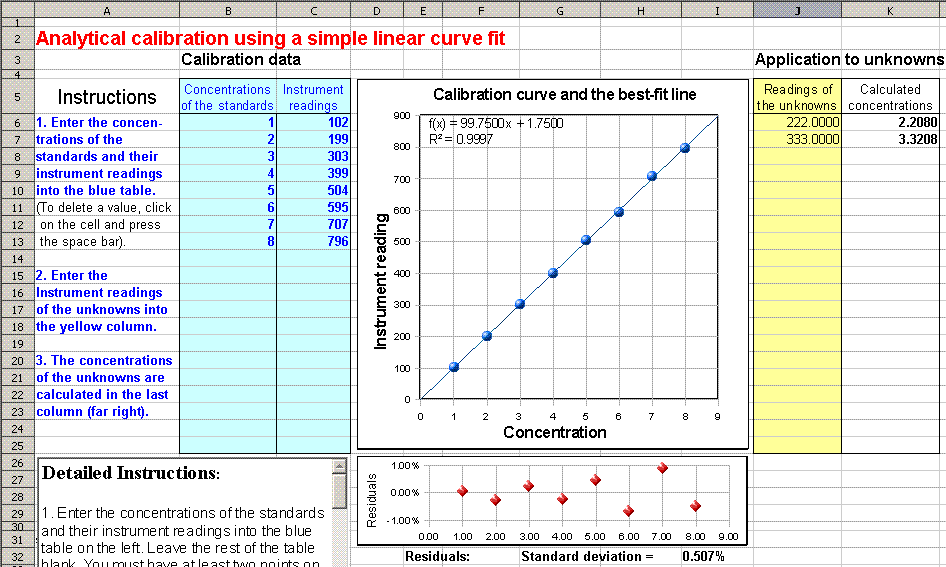
curve plot. (Click here for a
fill-in-the-blank OpenOffice spreadsheet that does this for
you. View
screen shot). If the residuals are randomly scattered,
then it means that the deviations are caused by random errors
such as photon or detector noise or random volumetric or
procedural errors. If the residuals have a smooth
shape, this means that the errors are systematic. If the
residual plot has a straight line segment at low
concentrations but curves off at high concentrations, then
it's probably stray light that is causing the non-linear
region.
25. What is the minimum value of the
coefficient of determination (R2) to obey
Beer's Law?
It depends on the accuracy required. As a rough rule of thumb,
if you need an accuracy of about 0.5%, you need an R2
of 0.9998; if a 1% error is good enough, an R2
of 0.997 will do; and if a 5% error is acceptable, an R2
of 0.97 will do. The bottom line is that the R2
must be pretty darned close to 1.0 for quantitative results in
analytical chemistry. But if the deviation from linearity is
smooth and gradual, rather than random, you can still get
accurate results with a "curvilinear" calibration curve
fitting technique, such as a quadratic
or cubic fit.
26. What does it mean if the
calibration curve (Beer's Law plot) does not extend
through zero?
If it's close to zero, it may simply be due to random error in
the absorbance readings or the volumetric preparations.
Another possibility is that the calibration curve shape is
curved, but you have fitted a straight line to it; use a non-linear curve fit
instead). Finally, it may mean that you have not
properly subtracted the "blank"
(see #16, above).
27. What is the effect of the slit
width on the spectra in uv-vis spectrophotometry?
The slit width determines the spectral bandpass, the
wavelength range of the light passing through the sample. The
smaller the slit width, the more nearly monochromatic the
light beam will be. But if the slit width is too large, the
polychromatic light effect will cause the spectral peaks to be
shorter and broader than they would be at narrower slit
widths. If you are trying to measure an accurate absorption
spectrum, for example for use as a reference spectrum for
future measurements or for identification of that absorber,
then you should use a narrow slit. However, the
signal-to-noise ratio decreases as the slit width is reduced,
so it is not always practical to use the smallest slit width
possible. If the spectral bandpass is one tenth (1/10th)
of the spectral width (full width at half-maximum) of the
narrowest band in the spectrum, then the maximum peak height
error caused by polychromaticity will be less than 1%.
28. How does Beer's Law apply to atomic absorption
spectroscopy?
Beer's Law is the basis of atomic absorption spectroscopy, as
it is for conventional molecular spectrophotometry. But there
is a big difference. In atomic absorption, the absorbers are free atoms in the gas phase
in a high-temperature flame or graphite furnace atomizer, and
their absorption spectra consist of very narrow spectral
"lines", only about 0.003 nm in width (compared to a typical
molecule in solution that might have a spectral width of 50 -
100 nm or more). So in order for Beer's Law to be obeyed with
such an extremely narrow absorption, you would to use a light
beam with an even narrower spectral width, ideally much less
than 0.003 nm. Ordinary monochromators can not achieve a
spectral bandpass anywhere near that narrow, so a conventional
optical design is impossible. The problem is solved in two
distinctly different ways. The most common and least
expensive type of atomic absorption instrument uses
an atomic vapor lamp
as the primary light source, which emits the atomic line
spectrum of the element to be determined. A small
monochromator is used, positioned between the flame or
graphite furnace and the detector, but its function is only to
isolate one line from the line source and to reduce stray
light. The line width of the light source is typically about
0.001 nm or so, not very much less than the absorption line
width, so adherence to Beer's Law is not perfect, but it's
good enough at low concentrations. The disadvantage is
that you need to purchase a separate lamp for each element you
intend to measure. The other type of instrument uses a continuum
source, a special type of high-resolution spectrometer
called an "echelle" spectrometer, and a diode-array detector.
The advantage of this approach is the ease of switching from
one element to the next and the possibility of simultaneous
multi-element measurement. However, a continuum source
instrument is much more expensive.
29. Why is it important not to
have fingerprints on the cuvette?
Fingerprints absorb and scatter light slightly, even though
they might not be readily visible. So a cuvette with
fingerprints on it will give a slightly higher absorbance
reading that a clean one. Unless you compensate for this
by by using the same cuvette, with the same exact fingerprint,
for the blank
solution, and subtract the blank signal from the
samples, the measured concentration will be inaccurate.
This is especially important if the absorbance is low
(say, below 0.01 absorbance).
30. Why is absorbance not measured
directly?
In absorption spectroscopy, the intensity of the
absorbed light can not measured directly because the absorbed
light is converted into heat, but the resulting temperature
increase is far too small to be readily measured without very
specialized and expensive equipment. The only
thing that can be measured directly is the intensity of the
transmitted beam. Making a calibration curve based on the intensity
of the transmitted beam result not a good idea because the
relationship to concentration is highly non-linear.






{kind=link}
{kind=link}
{kind=link}