Real-time simulation of a scanning fluorescence
spectrofluorometer. Students can set the excitation and emission
wavelengths, scan excitation spectra,
emission spectra, or synchronous spectra, change the
concentrations of
two fluorescent components, insert and remove the blank and sample
cuvettes, measure the wavelengths of maximum excitation and
emission, Stokes shift, and detection limits, observe Raleigh and
Raman scatter, dark current, photon noise, determine the
frequency of the vibration causing the Raman peak, compare
absorption to fluorescence measurement of
the same solution, optimize measurement of two-component mixture
by selective excitation and synchronous fluorescence methods,
generate and plot analytical curves automatically, and observe the
non-linearity and spectral distortion caused by self-absorption.
Download links: OpenOffice
Version, FluorescenceOO.ods Note: to run this spreadsheet,
you have to first download the OpenOffice installer (download from OpenOffice),
then install it (by double-clicking on the installer file that you
just
downloaded), and then download my spreadsheets from this page.
Once OpenOffice is installed, you can run my spreadsheets
just by
double-clicking on them. Note
1:Don't use version
3.1.
There is a bug in OpenOffice 3.1 that causes bad x-axis scaling on
some of my graphs. The problems does not occur in version
3.0 or in the most recent version 3.2. Note 2: Downloading these
files with Interent Explorer
will change the file types from ".ods" to ".zip"; you will have to
edit
the file names and change the extensions back to ".ods" for them
to
work properly. This problem does not occur in Firefox or in Chrome.
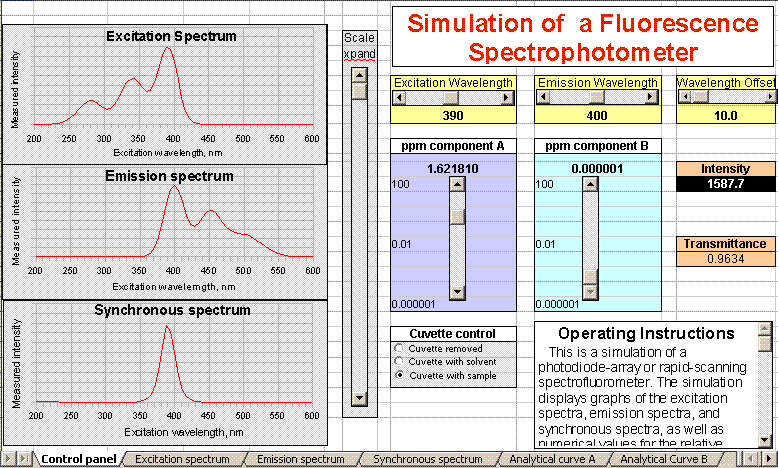
This is a simulation of a photodiode-array or rapid-scanning
spectrofluorometer. The simulation displays graphs of the excitation
spectra, emission spectra, and synchronous spectra, as well as
a numerical value for the relative fluorescence emission intensity
(the black box labeled "Intensity").
To change the excitation and emission wavelengths (in nm), adjust
the two sliders at the top, labeled "Excitation Wavelength" and "Emission Wavelength". To change
the graph scale expansion, use the long vertical slider labeled "Scale xpand". (This
effects only the vertical intensity scale of the graphs, not the
numerical intensity display).
To view these spectra enlarged, click the tabs at the bottom left of
the window, labeled Control,
Excitation, Emission, and Synchronous.
The sample solution may contain 1 or 2 fluorescent compounds, A and
B,
dissolved in water. You can control the concentrations of these two
components by using the sliders labeled "ppm component A" and "ppm component B". You can also control what is
placed in the sample compartment with the "Cuvette control"
buttons.
The
transmittance of the sample solution at the excitation wavelength is
also displayed, to aid in comparing fluorescence to absorption
measurement and as an indication of the extend of the
self-absorption
or "inner filter" effect.
The synchronous fluorescence spectrum is displayed at the bottom
left. The Wavelength Offset
control (top right) controls the wavelength difference between the
excitation and emission wavelengths in nm.
To inspect the analytical curve with respect to component A
alone (at constant concentration of component B), click the tab at
the
bottom of the windows labeled "Analytical curve A". To inspect
the analytical curve with respect to component B alone (at
constant concentration of component A), click the tab at the bottom of
the windows labeled "Analytical curve B".
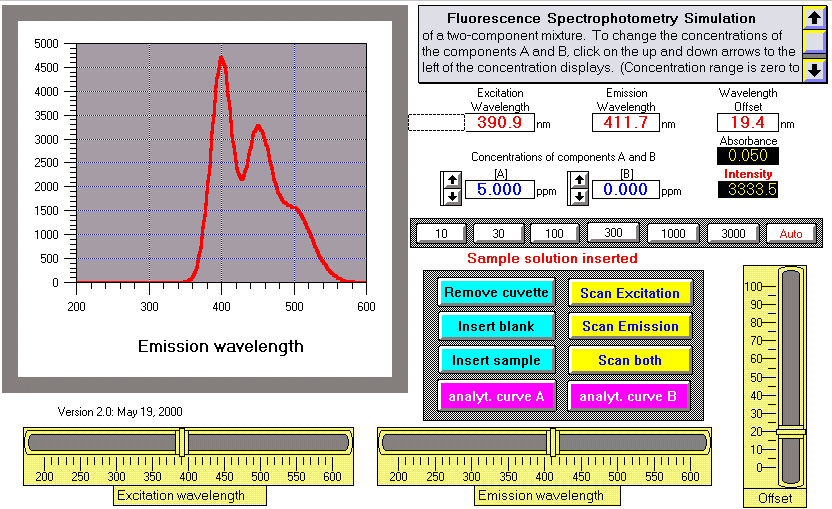
Operating instructions, WingZ
version:
To change the concentrations of the components A and B, click on
the up and down arrows to the left of the concentration
displays(concentration range is zero to 100 ppm in a 1,2, 5, 10
sequence); or you can type in any arbitrary concentration for
either
component while the cuvette is removed.
Fluorescence intensity (in arbitrary units) and the absorbance of
the
solution at the excitation wavelength are displayed in the black
boxes.
Readings are continuous as long as the cuvette is inserted into
the
instrument. (The random fluctuations in readings are due to photon
noise).
Clicking "Remove cuvette" simulates removal of the cuvette from
the
light path; the intensity read-out displays only the detector's
dark
current. Clicking "Insert blank" simulates inserting a cuvette
filled
with pure water into the light path; the intensity read-out
displays
the light scatter (Rayleigh and Raman) from the water. Clicking
"Insert
sample" simulates inserting a cuvette filled with a water solution
of
the two components at the specified concentrations. The cuvette
must be
removed to type in arbitrary concentrations and then inserted to
measure.
To change the excitation and emission wavelengths (in nm),
adjust the two sliders at the bottom. To scan a spectrum, click on
the
corresponding scan button. To obtain a synchronous spectrum, set
the
wavelength offset with the slider on the right and click "Scan
both".
Change the y-axis scale of the plots by clicking on one of the
seven
small "sensitivity" buttons labeled "10" through "3000", or press
"auto" to allow the computer to automatically adjust the y-axis
scale.
Note: the intensity and absorbance displays respond immediately to
changes in concentrations and wavelengths; however, spectra must
be
re-scanned after changing the concentrations, wavelengths, or
offset.
Pressing "analyt.curve A" runs an analytical curve for
component A and displays a log-log plot of intensity vs
concentration
of A from 0.001 to 100 ppm. Pressing "analyt. curve B" does the
same
thing for component B. Scanning a spectrum replaces the analytical
curve plot. From this plot it is possible to convert the relative
intensity readings into concentration in ppm.
Instructor's Notes:
This is a simulation of room temperature prompt fluorescence of two
non-interacting fluorophors in aqueous solution with right angle
geometry in a standard cuvette, measured with a corrected dispersive
spectrofluorometer. You can think of the two components as
two analytes or as one analyte (A) and a background or interfering
component (B). Both
components obey Vavilov's law (shapes of the emission spectra of
each
component separately are independent of the excitation wavelength,
and
vice versa, except for the scatter peaks). The simulation includes
Rayleigh and Raman scatter peaks of the solvent (water); there is
only
one Raman band observable, that of the OH stretch of water. (The
Rayleigh peak is fixed in
amplitude but the Raman band height varies with the inverse 4th
power
of wavelength). The Raman peak of water is often used to measure
the sensitivity and signal-to-noise ratio of fluorescence
instruments.
The
simulation includes the self-absorption (inner-filter) effect for
both the excitation beam
and the fluorescence emission, and it includes photon noise but not
flicker or detector (dark current) noise. In addition to an
intensity display, there is also an absorbance readout,
which gives the absorbance of the sample solution at the excitation
wavelength;
this is intended to allow a comparison of fluorescence to absorption
measurement
and as an indicator of the presence of self-absorption, but it is
not a full simulation of absorption spectrophotometric measurement
(it does not include
stray light or finite spectra bandwidth deviations nor background
shifts due to changes in cell transmission). The simulation has a
synchronous scanning mode (constant delta-lambda).
On most computers the scanning speed of the simulated instrument
will be faster than typical
real instruments.
There are several parameters that you can change, to modify the
simulation experience
for specific purposes. You can change the spectral characteristics
of
the two components. The excitation and the emission spectra are
each
modeled as three overlapping Gaussian bands. The
heights, peak wavelengths, and widths of each band are given in
the
table at R20..R46. For example, h1ax is the height of the
first band of component A's excitation spectrum, and
w3bm is the width of the third band of component B's
emission
spectrum, and so forth
(peak wavelengths and widths are in nm; height is in arbitrary
units).
You can change the overall signal-to-noise ratio of the instrument
(cell Q17). You can also change the sequence of concentrations
used to construct analytical curves (table in U10..U26 in the WingZ version and the large
table starting at U8 in the OpenOffice
version).
After making any changes, I suggest that you Save
the simulation under a different file name, so you preserve the
original.
Cell definitions and equations (forWingZ version 2.1):
Inputs:
Concentration of A in ppm (cell I12)
Concentration of B in ppm (cell K12)
ex = wavelength of excitation monochromator (cell I8 or excitation slider)
em = wavelength of emission monochromator (cell K8 or emission slider)
of = synchronous offset (cell M8 or offset slider)
epsa = absorption coefficient of component A
epsb = absorption coefficient of component B
snr = signal-to-noise ratio (Cell Q17)
Z1 = 1 if cuvette is inserted; 0 if removed from the instrument.
Excitation band characteristics of component A: (cells R20..R28)
band # 1 2 3
Height: h1ax h2ax h3ax
Position: p1ax p2ax p3ax
Width: w1ax w2ax w3ax
Emission band characteristics of component A: (cells R20..R28)
band # 1 2 3
Height: h1am h2am h3am
Position: p1am p2am p3am
Width: w1am w2am w3am
Excitation band characteristics of component B: (cells R29..R37)
band # 1 2 3
Height: h1bx h2bx h3bx
Position: p1bx p2bx p3bx
Width: w1bx w2bx w3bx
Emission band characteristics of component B: (cells R38..R46)
band # 1 2 3
Height: h1bm h2bm h3bm
Position: p1bm p2bm p3bm
Width: w1bm w2bm w3bm
U10..U26: sequence of component concentrations (ppm) for analytical curves.
Calculated quantities:
Concentration of A in ppb = A = 1000*ppmA
Concentration of B in ppb = B = 1000*ppmB
Wavelength of Raman peak in emission spectrum = raman = ex/(1-ex*0.00034)
Wavelength of Raman peak in excitation spectrum = xraman = em/(1-em*0.00034)
Intensity of Raman peak in emission spectrum = RamInt = 200000000000/ex^4
Intensity of Raman peak in excitation spectrum = xRamInt = 200000000000/em^4
Emission factor, component A
ema = (h1am*exp(-((em-p1am)/w1am)^2)
+h2am*exp(-((em-p2am)/w2am)^2)
+h3am*exp(-((em-p3am)/w3am)^2))
Emission factor, component B
emb = (h1bm*exp(-((em-p1bm)/w1bm)^2)
+h2bm*exp(-((em-p2bm)/w2bm)^2)
+h3bm*exp(-((em-p3bm)/w3bm)^2))
Excitation factor, component A
exa = (h1ax*exp(-((ex-p1ax)/w1ax)^2)
+h2ax*exp(-((ex-p2ax)/w2ax)^2)
+h3ax*exp(-((ex-p3ax)/w3ax)^2))
Excitation factor, component B
exb = (h1bx*exp(-((ex-p1bx)/w1bx)^2)
+h2bx*exp(-((ex-p2bx)/w2bx)^2)
+h3bx*exp(-((ex-p3bx)/w3bx)^2))
Absorbance of sample solution at the excitation wavelength
Aex = epsa*A*(h1ax*exp(-((ex-p1ax)/w1ax)^2)
+h2ax*exp(-((ex-p2ax)/w2ax)^2)
+h3ax*exp(-((ex-p3ax)/w2ax)^2))
+epsb*B*(h1bx*exp(-((ex-p1bx)/w1bx)^2)
+h2bx*exp(-((ex-p2bx)/w2bx)^2)
+h3bx*exp(-((ex-p3bx)/w3bx)^2))
Absorbance of sample solution at the emission wavelength
Aem = epsa*A*(h1ax*exp(-((em-p1ax)/w1ax)^2)
+h2ax*exp(-((em-p2ax)/w2ax)^2)
+h3ax*exp(-((em-p3ax)/w2ax)^2))
+epsb*B*(h1bx*exp(-((em-p1bx)/w1bx)^2)
+h2bx*exp(-((em-p2bx)/w2bx)^2)
+h3bx*exp(-((em-p3bx)/w3bx)^2))
Transmission of sample solution at the excitation wavelength
Tex = 10^(-Aex)
Transmission of sample solution at the emission wavelength
Tem = 10^(-Aem)
Total output intensity (fluorscence + scatter + Raman) (cell M13)
total = Z1*Tex*Tem*((A*ema*exa+B*emb*exb)
+100*exp(-((ex-em)/10)^2)
+RamInt*exp(-((em-raman)/10)^2))
Display outputs:
Absorbance (cell M20)
= Aex + 0.001*(rand()-0.5)
Intensity (cell M12)
=abs(total+(sqrt(total)+2)*(rand())/snr)
Array calculations:
D31..D101: wavelength, 200..600 nm in 6 nm steps
B31..B101: absorbance of solution at wavelength
absorbance = epsa*A*(h1ax*exp(-((wavelength-p1ax)/w1ax)^2)
+h2ax*exp(-((wavelength-p2ax)/w2ax)^2)
+h3ax*exp(-((wavelength-p3ax)/w3ax)^2))
+epsb*B*(h1bx*exp(-((wavelength-p1ax)/w1bx)^2)
+h2bx*exp(-((wavelength-p2ax)/w2bx)^2)
+h3bx*exp(-((wavelength-p3ax)/w3bx)^2))
C31..C101: transmission of solution at wavelength
transmission = 10^(absorbance)
E31..E101: excitation spectrum (including Rayleigh and Raman scatter)
excitation = Tem*transmission*(A*((h1ax*exp(-((wavelength-p1ax)/w1ax)^2)
+h2ax*exp(-((wavelength-p2ax)/w2ax)^2)
+h3ax*exp(-((wavelength-p3ax)/w3ax)^2))*ema)
+B*((h1bx*exp(-((wavelength-p1bx)/w1bx)^2)
+h2bx*exp(-((wavelength-p2bx)/w2bx)^2)
+h3bx*exp(-((wavelength-p3bx)/w3bx)^2))*emb)
+100*exp(-((wavelength-em)/10)^2)
+xRamInt*exp(-((wavelength-xraman)/10)^2))
G31..G101: excitation spectrum with photon noise
ex+noise = $Z$1*(abs(excitation+(sqrt(excitation)+2)*(rand())/snr))
I31..I101: emission spectrum (including Rayleigh and Raman scatter)
emission = Tex*transmission*(A*(exa*(h1am*exp(-((wavelength-p1am)/w1am)^2)
+h2am*exp(-((wavelength-p2am)/w2am)^2)
+h3am*exp(-((wavelength-p3am)/w3am)^2)))
+B*(exb*(h1bm*exp(-((wavelength-p1bm)/w1bm)^2)
+h2bm*exp(-((wavelength-p2bm)/w2bm)^2)
+h3bm*exp(-((wavelength-p3bm)/w3bm)^2)))
+100*exp(-((wavelength-ex)/10)^2)
+RamInt*exp(-((wavelength-raman)/10)^2))
K31..K101: emission spectrum with photon noise
em+noise = $Z$1*(abs(emission+(sqrt(emission)+2)*(rand())/snr))
Transmission at offset wavelength (wavelength+offset)
A31..A101: Toff
Toff = 10^(-epsa*A*(h1ax*exp(-((wavelength-p1ax+of)/w1ax)^2)
+h2ax*exp(-((wavelength-p2ax+of)/w2ax)^2)
+h3ax*exp(-((wavelength-p3ax+of)/w3ax)^2))
+epsb*B*(h1bx*exp(-((wavelength-p1ax+of)/w1bx)^2)
+h2bx*exp(-((wavelength-p2ax+of)/w2bx)^2)
+h3bx*exp(-((wavelength-p3ax+of)/w3bx)^2)))
M31..M101: synchronous spectrum (including Rayleigh and Raman scatter)
synch = Toff*transmission*(A*((h1ax*exp(-((wavelength-p1ax)/w1ax)^2)
+h2ax*exp(-((wavelength-p2ax)/w2ax)^2)
+h3ax*exp(-((wavelength-p3ax)/w3ax)^2))
*(h1am*exp(-((wavelength-p1am+of)/w1am)^2)
+h2am*exp(-((wavelength-p2am+of)/w2am)^2)
+h3am*exp(-((wavelength-p3am+of)/w3am)^2)))
+B*((h1bx*exp(-((wavelength-p1bx)/w1bx)^2)
+h2bx*exp(-((wavelength-p2bx)/w2bx)^2)
+h3bx*exp(-((wavelength-p3bx)/w3bx)^2))
*(h1bm*exp(-((wavelength-p1bm+of)/w1bm)^2)
+h2bm*exp(-((wavelength-p2bm+of)/w2bm)^2)
+h3bm*exp(-((wavelength-p3bm+of)/w3bm)^2)))
+100*exp(-((of)/10)^2)
+RamInt*exp(-((wavelength+of-(wavelength/(1-wavelength*0.00034)))/10)^2))
O31..O101: synchronous spectrum with photon noise
synch+noise = $Z$1*(abs(synch+(sqrt(synch)+2)*(rand()/snr)))
Graphs:
Excitation spectrum: excitation+noise vs excitation wavelength
Emission spectrum: emission+noise vs emission wavelength
Synchronous spectrum: sync+noise vs excitation wavelength
Analytical curves: Intensity vs concentration of A or B in ppm
Student assignment, WingZ version:
Simulation
of Scanning Fluorescence Spectrometer
This is a
simulation of a scanning spectrofluorometer. The simulation
displays excitation spectra, emission spectra, and synchronous
spectra, relative fluorescence intensity, and absorbance at the excitation
wavelength. Operating instructions are contained in the
scrolling text field in the upper right of the screen.
Answer the following questions on a separate sheet to turn
in. Please do not make repeated print-outs of
this spreadsheet.
1. Set A=1 ppm
and B=0. Determine the wavelengths of maximum excitation and
emission for component A. What is its Stokes shift?
2. Does Vavilov's
Law* seem to hold for compound A, that is, is the shape of the
emission spectra independent of the excitation wavelength, and vice
versa, except for the scatter peaks?
3. Is there any
sign of Rayleigh or Raman scatter? How could you distinguish these
from
genuine fluorescence?
4. Check the
blank (click on "Insert Blank"). Increase the sensitivity setting
as necessary. Is there any sign of dark current or background
fluorescence? What are the main features of the excitation and emission
spectra of the blank. Estimate the spectral bandpass of the monochromators.
5. Does the
wavelength separation between the Rayleigh and Raman scatter peaks in the
emission spectrum vary with excitation wavelength? What is the
frequency, in cm-1, of the vibration causing the Raman peak? What
vibration is most likely the cause?
6. Find the
combination of excitation and emission wavelength that gives the best
precision of measurement of low concentrations of component A.
Estimate the detection limit of component A in ppm. Is the detection
limit lower by fluorescence or by absorption measurement? By approximately
what factor?
7. Over most of
the concentration range, what is the source of noise in the
intensity readings and in the spectra? How could you prove this?
8. Is there
evidence of non-linearity in the relationship between concentration and
intensity at high concentrations? What is the most likely source of the
non-linearity?
9. Vary the
wavelength offset and observe the synchronous spectrum. What offset gives
the largest peak height? Explain the effect of Rayleigh and Raman
scatter on the synchronous spectrum. Note: this is a constant
wavelength synchronous spectrum.
10. Set A=0 and
B=1 ppm. Determine the wavelengths of maximum excitation and emission for
component B. What is its Stokes shift? Can mixtures of these two
components be determined by fluorescence measurement?
*
Vavilov's Law states that the shape of the fluorescence emission
spectrum of a single fluorophor is independent of the excitation
wavelength, and vice versa. This holds only for the
fluorescence
peaks, not Raman or scatter peaks.
Frequently-Asked Questions
1. Question:Why is fluorescence intensity measured
in "arbitrary units"? Answer:
Nothing more is needed for the common applications of fluorescence
spectroscopy: for quantitative applications, a calibration curve can
be
prepared and used on the same instrument employing any set of
intensity
units equally well, and for qualitative applications, the shape of a
spectrum is the same no matter what intensity units are used.
Calibrating a fluorescence spectrophotometer to read in absolute
physical units (watts, for example) would add to the cost and would
not
result in any real advantage for most applications. Most
fluorescence
spectrophotometers simply measure the detector signal, which
is proportional to the fluorescence intensity. The magnitude of
that signal at a given analyte concentration is somewhat arbitrary,
depending upon the choice of detector and its operating conditions,
as
well as the intensity of the light source and the characteristics of
the monochromator and the rest of the optics. See "Signal-to-Noise Ratio and Detection
Limit of Fluorescence Spectroscopy" for a detailed analysis of
the factors influencing the detector signal.
2. Question: Why not measure absorbance or
transmission, as in absorption spectroscopy? Answer: This
is due to the different optical configuration of absorption and
fluorescence measurement. In absorption spectrometry, the light beam
that passes through the sample is measured directly by a detector
placed a 180º angle from the incident beam, which allows the
incident
beam intensity to be measured simply by replacing the sample with a
"blank". In fluorescence spectrometry, the light beam that
passes
through the sample is not measured at all; rather,
the fluorescence emission is measured, usually at a 90º
angle
from the incident beam. Therefore, absorbance or
transmission
can not be measured in a fluorescence spectrometer because the
detector does not measure the incident beam. Even if you tried to
move
the detector around to 180º to measure the incident beam, it
wouldn't work very well because the light intensity of the incident
beam can be as much as 1013 times brighter
than the fluorescence intensity, and no detector in common use can
measure intensities over that wide a range. (Note: some
instruments do sample a small portion of the incident bean and
measure
it with a second (lower sensitivity) detector, then calculate the
ratio
of the fluorescence intensity to the incident intensity. Even
this ratio, however, is not comparable between instruments, in the
same
way that absorbances can be compared
3. Question: How does fluorescence intensity vary
with concentration? Answer: Fluorescence
intensity is directly proportional to concentration (i.e. the
calibration curve is linear), as long as the absorbance of the
analyte
at the excitation wavelengths is very low, say, less than about
0.004
(99% T). If the absorbance is greater than this, then the
excitation intensity is reduced by the absorption, and
the calibration curve becomes non-linear (concave down). The
effect is exacerbated if the analyte also absorbs at the emission
wavelength.
4. Question: Why is fluorescence measured at a 90º
angle to the incident (excitation) beam? Answer: There
is nothing magic about 90º. The fluorescence can't be
measured at a 180º, because the transmitted beam would completely
swamp
the fluorescence. The choice of 90º is a matter of
convenience,
especially when conventional square cross-section cuvettes are
used.
5. Question: What are the relative detection limits
of absorption and fluorescence measurement? Answer: This depends
largely on thefluorescence quantum efficiency
of the analyte (the ratio of the number of photons emitted as
fluorescence to the number absorbed). For a highly fluorescent
molecule
such as quinine, which has
a
quantum efficiency of 0.5, fluorescence has a better (lower)
theoretical detection limit than absorption by a factor of roughly
103 using comparable instrumentation, as
you can demonstrate by comparing AbsorptionSNR.ods
to FluorescenceSNR.ods.
But most fluorescent molecules have quantum efficiencies lower than
0.5, so the advantage of fluorescence is not so large on average.
But
by the same token, any molecule with a quantum efficiency higher
than
0.0005 is likely to be at least marginally better by fluorescence,
at
least in terms of theoretical detection limit.
6. Question: How can a fluorescent peak can be
distinguish from an Raman peak? Answer: Vary
the excitation wavelength slightly. If the peak shifts in wavelength
and its wavelength is longer than the excitation wavelength, it's a
Raman peak; if it changes its intensity but not its wavelength, it's a
fluorescent peak.
7. Question: Why choose a fluorescence measurement
over absorption? After all, any molecule that fluoresces must also absorb. Answer: The
choice depends on the fluorescence characteristics of the analyte
and
the matrix that it is present in. If the analyte exhibits
fluorescence
at excitation and emission wavelengths within the range of your
instrument, then it is a candidate for fluorescence measurement, if
its
quantum efficiency is high enough. But a further requirement is that
the fluorescence emissions of other chemical components in the
sample
not interfere, either because they are very weak or because the
excitation or emission wavelengths of those components can be
successfully resolved from that of the analyte. Each case has
to
be evaluated individually. Usually a fluorescence measurement is
chosen
over an absorption measurement when the analyte is strongly
fluorescent
and the other components in the sample have absorption that
interfere
with an absorption measurement but do not exhibit strong
fluorescence
emission that would interfere with a fluorescence measurement.
(c) 1991, 2015. This page is
part of Interactive
Computer Models for Analytical Chemistry Instruction, created
and maintained by Prof.
Tom O'Haver , Professor Emeritus, The University of Maryland
at College Park. Comments, suggestions and questions should be
directed to Prof. O'Haver at toh@umd.edu.
Number of unique visits since May 17, 2008: